

Note d’application Axcend – Analyse de produits pharmaceutiques par HPLC capillaire-MS/MS
ANALYSE DES PRODUITS PHARMACEUTIQUES A L’AIDE D’UN SYSTEME HPLC COMPACT
Cliquez ici pour en savoir plus sur la mini HPLC Axcend Focus LC.
S.V. Calugaru, E.P. Gates, W.R. West, M.L. Lee
Résumé
Cette note d’application démontre l’utilisation d’un chromatographe liquide capillaire portable compact, l’Axcend Focus LC, couplé à un spectromètre de masse triple quadripôle Agilent Ultivo pour l’analyse quantitative de médicaments pharmaceutiques dans des échantillons aqueux. Deux mélanges de test ont été analysés dans cette étude. Le mélange n°1 contenait de l’acétaminophène, de la caféine, de la carbamazépine, de la ciprofloxacine, de l’érythromycine, de la fluoxétine, du sulfaméthoxazole et du triméthoprime. Le mélange n°2 était composé de gemfibrozil, d’ibuprofène, de naproxène et de triclosan. Tous les composés des mélanges de test ont été identifiés et des courbes d’étalonnage ont été établies dans les plages de concentrations suivantes : 30-1 000 ng/mL pour la carbamazépine, 50-10 000 ng/mL pour le triméthoprime, 100-10 000 ng/mL pour l’acétaminophène, 100-5 000 ng/mL pour la caféine, 300-10 000 ng/mL pour la fluoxétine, 300-3 000 ng/mL pour le sulfaméthoxazole, 500-10 000 ng/mL pour l’érythromycine et la ciprofloxacine, 30-10 000 ng/mL pour le gemfibrozil, l’ibuprofène et le naproxène, et 30-1 000 ng/mL pour le triclosan.
Introduction
L’utilisation généralisée des produits pharmaceutiques, y compris les hormones, les antibiotiques, les anti-inflammatoires non stéroïdiens, les antidépresseurs et les antifongiques, entraîne un rejet important de ces médicaments et de leurs métabolites dans les eaux usées [1]. Des préoccupations ont été soulevées concernant leur potentiel à contaminer les eaux souterraines et superficielles et à pénétrer dans les approvisionnements en eau potable. Par conséquent, la surveillance des produits pharmaceutiques dans les échantillons aqueux représente une tâche analytique importante. L’utilisation de la chromatographie liquide capillaire à cette fin présente de nombreux avantages, notamment une consommation considérablement réduite de solvants organiques toxiques et coûteux [2] et une sensibilité potentiellement plus élevée lors de l’utilisation de la détection par spectrométrie de masse (MS) grâce aux faibles débits intrinsèques de la phase mobile [3].
Détails expérimentaux
Instrumentation
Un chromatographe liquide capillaire compact, Axcend Focus LC avec le logiciel Axcend Drive 2.2.0 (Axcend, Provo, Utah, USA), a été utilisé dans cette étude. Le système LC était connecté à un spectromètre de masse triple quadripôle, l’Ultivo LC/TQ G6465B équipé de la source d’ionisation par électrospray Jet Stream (Agilent Technologies, Santa Clara, Californie, USA). Le logiciel MassHunter (Acquisition : v1.1, Analyse qualitative : v10.0, Analyse quantitative : v10.0) a été utilisé pour le contrôle du spectromètre de masse et le traitement des données. Pour s’adapter aux débits micro de la phase mobile du LC capillaire, le nébuliseur standard de la source d’ionisation a été remplacé par un nébuliseur microflow (Numéro de pièce G1946-67260) obtenu auprès d’Agilent. Un tube PEEKsil de 25 cm de long, 360 µm OD, 25 µm ID (Numéro de pièce 0624374, Trajan, Melbourne, Victoria, Australie) a servi de ligne de transfert. Une extrémité de ce tube a été raccordée à la sortie du solvant de la cartouche de colonne à l’aide d’une union PEEK à zéro volume mort 360 µm avec un trou de 50 µm (Numéro de pièce C360UPK2, VICI Valco Instruments, Houston, Texas, USA). L’autre extrémité du tube a été connectée au nébuliseur microflow à l’aide d’un union réducteur en acier inoxydable 1/16″ à zéro volume mort 360 µm avec un trou de 100 µm (Numéro de pièce C360RUS64, VICI Valco Instruments). L’infusion directe de l’échantillon dans le spectromètre de masse pour l’optimisation des paramètres d’acquisition a été réalisée avec une pompe à seringue Modèle 22 de Harvard Apparatus (Holliston, Massachusetts, USA).
Réactifs et solvants
Les mélanges de produits pharmaceutiques (mélange n°1 contenant de l’acétaminophène, de la caféine, de la carbamazépine, de la ciprofloxacine, de l’érythromycine, de la fluoxétine, du sulfaméthoxazole et du triméthoprime à 200 µg/mL chacun dans du méthanol, et mélange n°2 contenant du gemfibrozil, de l’ibuprofène, du naproxène et du triclosan à 200 µg/mL chacun dans du méthanol) ont été achetés chez Restek (Bellefonte, Pennsylvanie, USA). Les étalons de calibration ont été préparés par dilutions en série de ces mélanges de test dans de l’eau. L’acétaminophène D4 (100 µg/mL dans du méthanol) et la carbamazépine D10 (100 µg/mL dans du méthanol) ont été achetés chez HPC Standards (Atlanta, Géorgie, USA) et utilisés comme étalons internes dans les échantillons du Mélange n°1. L’ibuprofène D3 (100 µg/mL dans du méthanol) a été acheté chez Cerilliant (Round Rock, Texas, USA) et utilisé comme étalon interne dans les échantillons du Mélange n°2. Les solvants de qualité LC-MS (eau et acétonitrile) ont été obtenus chez Sigma-Aldrich (MilliporeSigma, St. Louis, Missouri, USA) et l’acide formique de qualité LC-MS a été acheté chez ThermoFisher Scientific (Waltham, Massachusetts, USA).
Méthodes HPLC
Le mélange n°1 a été analysé en utilisant une colonne Acquity UPLC HSS T3 (10 cm x 150 µm i.d., taille de particules 1,8 µm) de Waters (Milford, Massachusetts, USA). Un détecteur à cellule de flux de petit diamètre a été utilisé pour mesurer l’absorption UV à 275 nm. Le mélange n°2 a été analysé en utilisant une colonne C18 (10 cm x 150 µm i.d., taille de particules 1,7 µm) de CoAnn Technologies (Richland, Washington, USA). L’absorption UV a été mesurée à 235 nm à l’aide d’un détecteur sur colonne. Dans les deux méthodes LC, le solvant A était composé de 97 % d’eau, 3 % d’acétonitrile et 0,1 % d’acide formique, et le solvant B était composé de 97 % d’acétonitrile, 3 % d’eau et 0,1 % d’acide formique. Le débit était de 1 µL/min et le volume d’injection était de 250 nL (boucle complète). Pour la séparation des composants du mélange n°1, le contenu du solvant B dans la phase mobile était de 3 % pendant les 0,5 premières minutes après l’injection de l’échantillon, puis il a été augmenté linéairement à 26 % pendant 1,5 min, maintenu à 26 % pendant 5 min, augmenté linéairement à 97 % pendant les 3 min suivantes, et finalement maintenu à 97 % pendant 5 min. Pour le Mélange n°2, le contenu du solvant B dans le gradient linéaire de phase mobile était de 3 % pour les 0,5 premières minutes, puis il a été augmenté à 60 % en 1 min, à 77 % pendant les 2,5 min suivantes, à 97 % pendant les 1,5 min suivantes, et enfin il a été maintenu à 97 % pendant 3 min.
Méthodes MS
La détection par réaction multiple (MRM) a été utilisée en mode de polarité positive et négative pour l’analyse des deux mélanges de test. L’acétaminophène deutéré, la carbamazépine et l’ibuprofène ont été utilisés comme étalons internes. Les méthodes MRM ont été optimisées à l’aide du logiciel MassHunter optimizer en injectant le mélange de test dilué respectif (chaque composant à 100 ng/mL) dans la source d’ions à un débit de 1 µL/min à l’aide d’une pompe à seringue. Les valeurs optimales de tension de et d’énergie de collision pour les ions quantificateurs et qualificatifs des analytes individuels et des étalons internes sont présentées dans le Tableau 1 pour le Mélange n°1 et dans le Tableau 2 pour le Mélange n°2. Les autres paramètres MS étaient les suivants : tension de la capillaire, 3000 V ; température du gaz, 200 °C ; débit du gaz, 5 L/min ; pression du nébuliseur, 10 psi ; et temps de résidence, 50 ms, sauf pour la fluoxétine et le sulfaméthoxazole, pour lesquels il était de 100 ms.
Résultats et discussion
Identification des analytes
Deux transitions entre l’ion parent et les ions produits ont été sélectionnées pour la surveillance des analytes et des étalons internes. La première transition était utilisée pour la quantification de l’analyte et la deuxième pour la qualification (Tableaux 1 et 2). Aucune fragmentation du triclosan n’a été observée dans les conditions testées. Pour ce médicament, seul l’ion moléculaire a été surveillé (Tableau 2). Les structures chimiques des produits pharmaceutiques analysés sont montrées dans la Figure 1 pour le mélange n°1 et dans la Figure 2 pour le mélange n°2. Les séparations chromatographiques des composants du Mélange n°1 et du Mélange n°2 avec détection UV à 275 nm et 235 nm sont présentées dans les Figures 3 et 4, respectivement. Chaque analyte dans les chromatogrammes UV a été identifié en fonction des données MS. Les chromatogrammes des ions extraits pour le mélange n°1 et le mélange n°2 sont montrés dans les Figures 5 et 6, respectivement.
Quantification des analytes
Pour normaliser les intensités des signaux MS des composants du Mélange n°1, des analogues deutérés de l’acétaminophène (D4-acétaminophène) et de la carbamazépine (D10-carbamazépine) ont été utilisés comme étalons internes à une concentration finale de 500 ng/mL chacun. Le D4-acétaminophène a servi d’étalon interne pour l’acétaminophène et la ciprofloxacine, tandis que le D10-carbamazépine a été utilisé pour les autres composés du mélange n°1. Des courbes de calibration linéaires avec un coefficient de régression R² > 0,99 ont été obtenues dans les plages de concentrations suivantes : 30-1 000 ng/mL pour la carbamazépine, 50-10 000 ng/mL pour le triméthoprime, 100-10 000 ng/mL pour l’acétaminophène, 100-5 000 ng/mL pour la caféine, 300-10 000 ng/mL pour la fluoxétine, 300-3 000 ng/mL pour le sulfaméthoxazole et 500-10 000 ng/mL pour l’érythromycine. La régression quadratique a été utilisée pour obtenir des courbes de calibration avec un coefficient de régression R² > 0,99 dans la plage de concentrations de 500-10 000 ng/mL pour la ciprofloxacine. Des courbes de calibration typiques sont montrées dans les Figures 7 et 8 pour l’acétaminophène et la carbamazépine, respectivement. Dans le cas du mélange n°2, le D3-ibuprofène a été utilisé à une concentration finale de 250 ng/mL comme étalon interne pour tous les analytes. Des courbes de calibration avec un coefficient de régression R² > 0,99 ont été obtenues dans les plages de concentrations suivantes : 30-10 000 ng/mL pour le gemfibrozil, l’ibuprofène et le naproxène (régression linéaire), et 30-1 000 ng/mL pour le triclosan (régression quadratique). Des courbes de calibration représentatives sont montrées dans les Figures 9 et 10 pour l’ibuprofène et le naproxène, respectivement.
Les résultats de cette étude peuvent être utilisés dans le développement de méthodes quantitatives pour l’analyse des produits pharmaceutiques dans des matrices complexes. Les méthodes HPLC-MS/MS présentées peuvent être appliquées, par exemple, à l’analyse de diverses fractions dans le cadre du développement et de l’optimisation des procédures de préparation des échantillons.
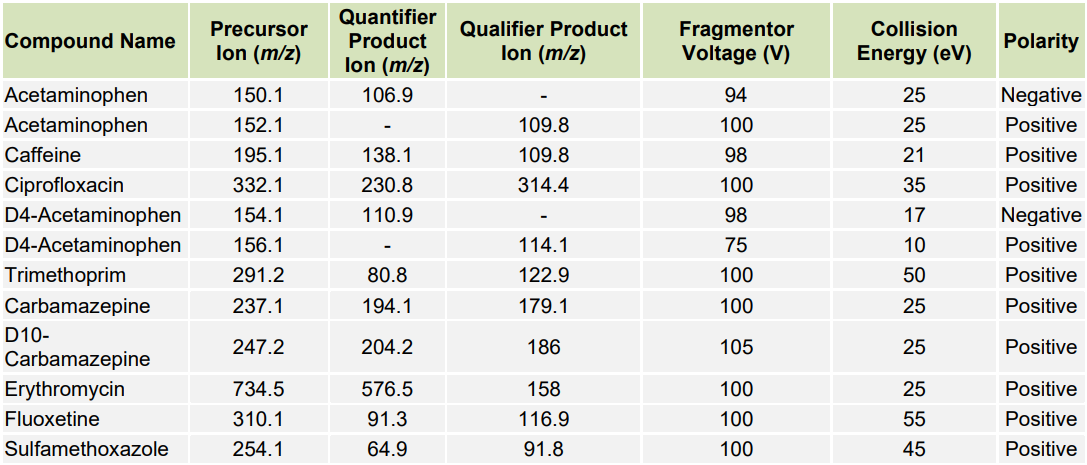
Table 1. Paramètres d’acquisition optimisés pour la détection MS des ions précurseurs et des ions produits des composés dans le mélange n°1
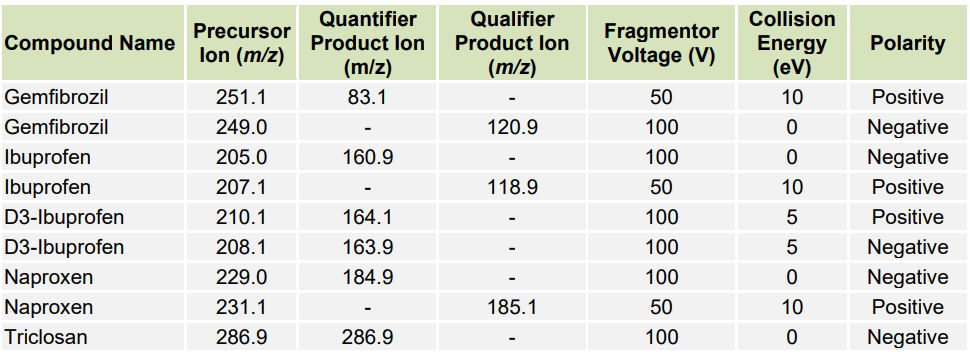
Table 2. Paramètres d’acquisition optimisés pour la détection MS des ions précurseurs et des ions produits des composés dans le mélange n°2
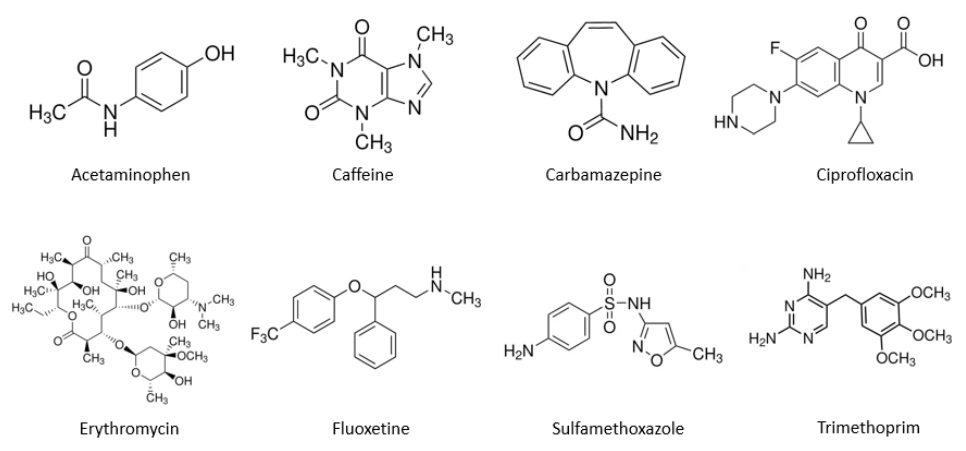
Figure 1. Structures chimiques des composés du mélange n°1
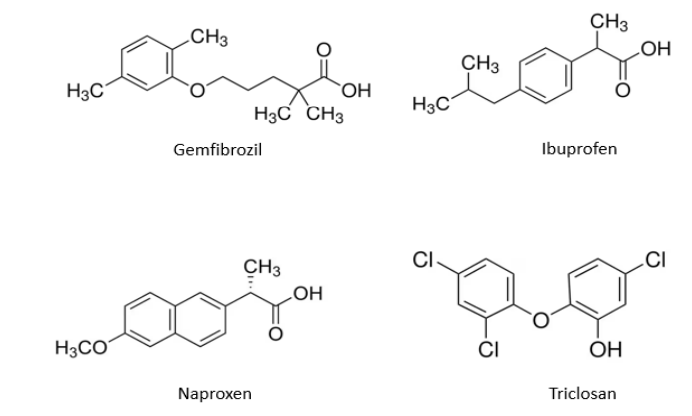
Figure 2. Structures chimiques des composés du mélange n°2
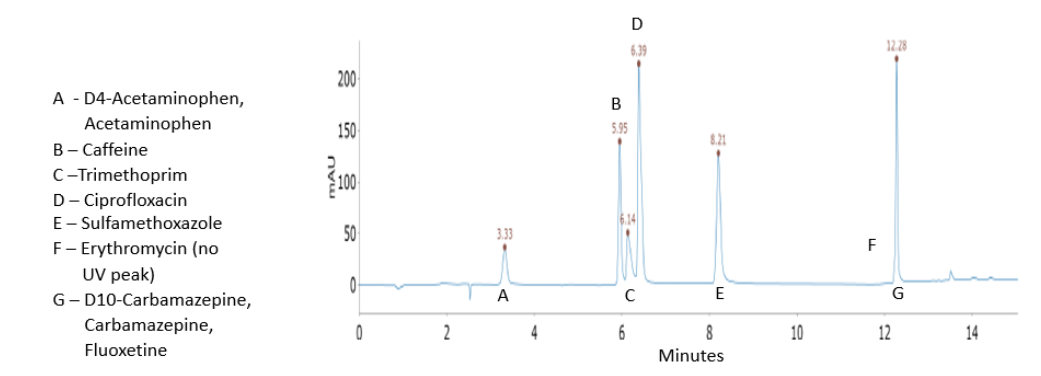
Figure 3. Analyse du mélange n°1 (5 µg/mL pour chaque composé) avec détection UV à 275 nm. Les identifications sont basées sur les données MS (voir Figure 5).
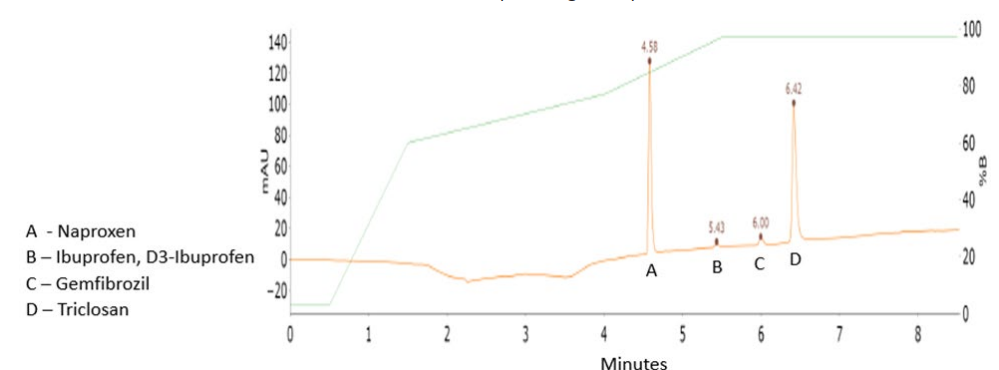
Figure 4. Analyse du mélange n°2 (10 µg/mL pour chaque composé) avec détection UV à 235 nm. Les identifications sont basées sur les données MS (voir Figure 6).

Figure 5. Analyse du mélange n°1 (5 µg/mL pour chaque composé) avec détection MS en mode MRM: TIC en haut et EIC en bas.
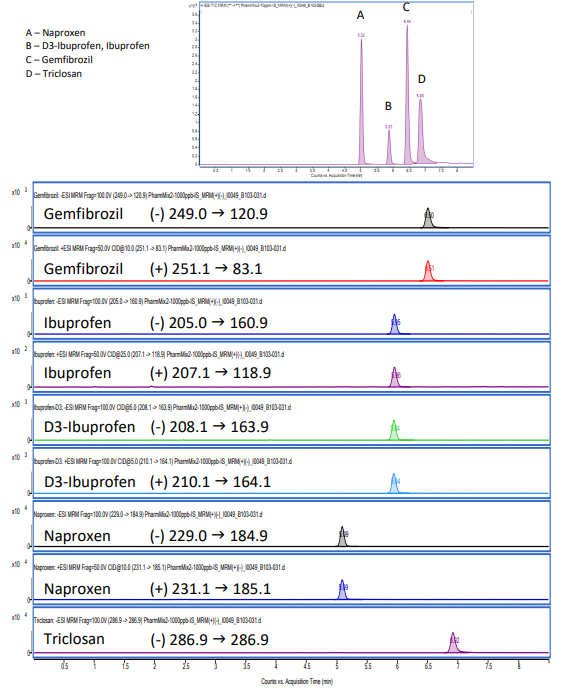
Figure 6. Analyse du mélange n°1 (10 µg/mL pour chaque composé) avec détection MS en mode MRM: TIC en haut et EIC en bas.
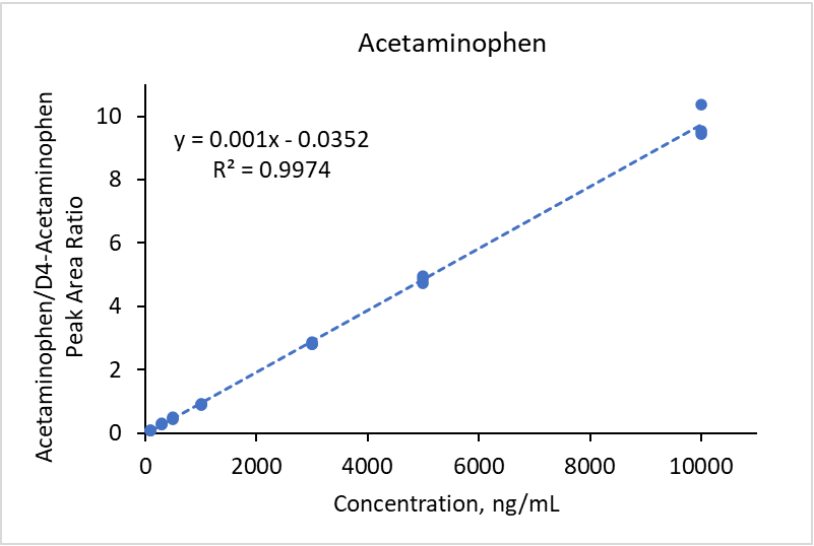
Figure 7. Courbe de calibration pour l’acétaminophène dans l’intervalle de concentration 100 -10000 ng/mL basée sur l’aires des pics.
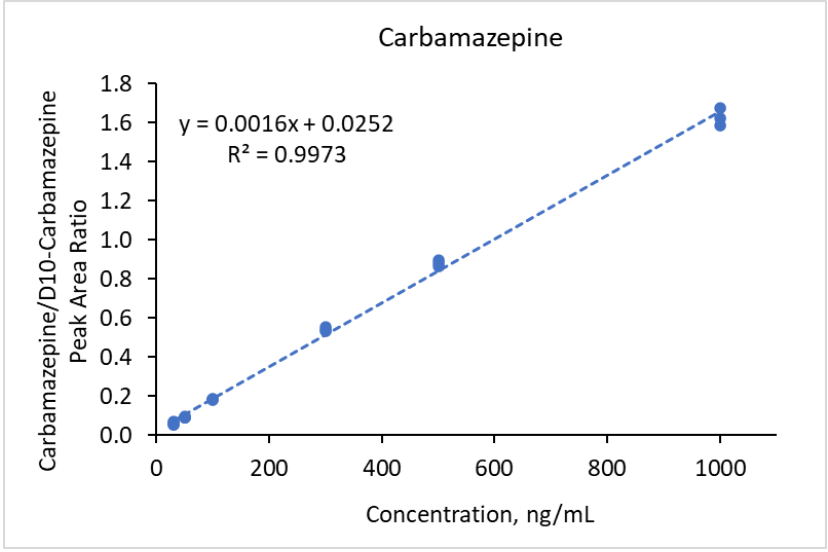
Figure 8. Courbe de calibration pour la carbamazépine dans l’intervalle de concentration 30 -1000 ng/mL basée sur l’aires des pics.
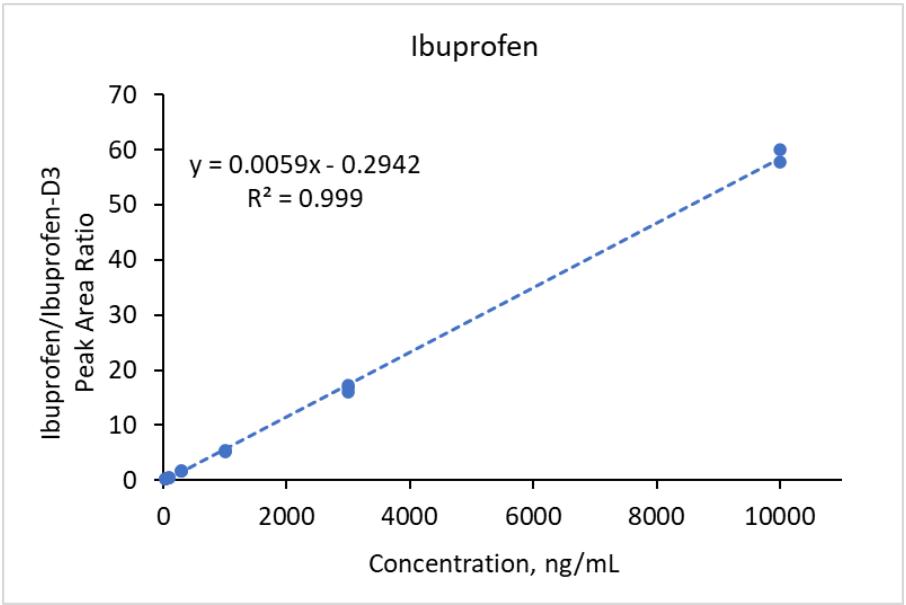
Figure 9. Courbes de calibration pour l’ibuprofène dans l’intervalle de concentration 30 -10000 ng/mL basée sur l’aires des pics.
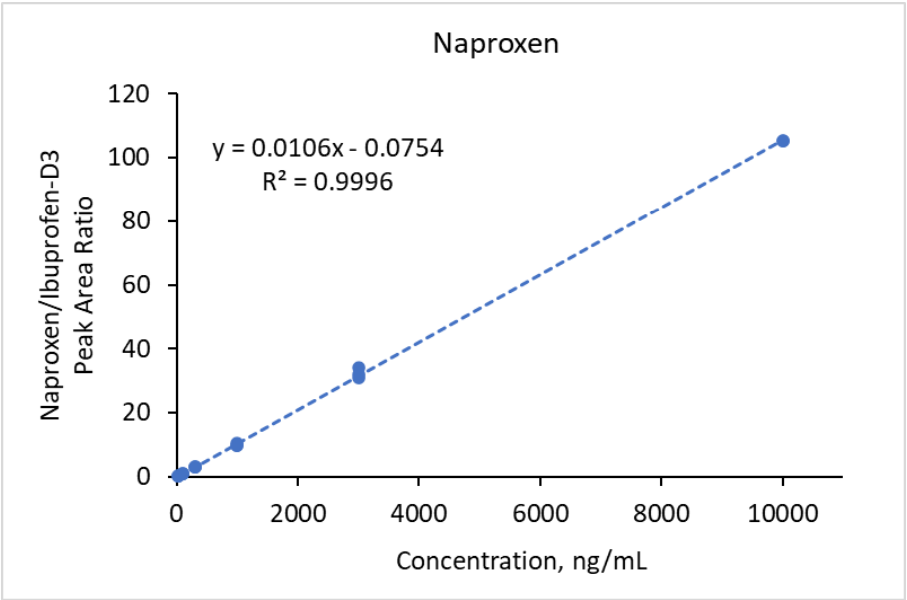
Figure 9. Courbe de calibration pour le naproxène dans l’intervalle de concentration 30 -10000 ng/mL basée sur l’aires des pics.
Conclusion
L’identification et la quantification des produits pharmaceutiques dans des mélanges de composants dans des échantillons aqueux modèles, basées sur la HPLC capillaire couplée à la spectrométrie de masse en tandem, ont été réalisées avec succès.
Des courbes de calibration avec un coefficient de régression R² supérieur à 0,99 ont été obtenues dans des plages de concentrations couvrant généralement au moins deux ordres de grandeur (par exemple, 100-10 000 ng/mL pour l’acétaminophène).
Les méthodes HPLC-MS/MS développées peuvent servir de base pour le développement ultérieur de la détermination quantitative des produits pharmaceutiques dans les matrices biologiques par HPLC-MS/MS capillaire.
Cliquez ici pour en savoir plus sur la mini HPLC Axcend Focus LC.
Références
- M.R. Letsoalo, T. Sithole, S. Mufamadi, Z. Mazhandu, M. Sillanpaa, A. Kaushik, T. Mashifana. Efficient Detection and Treatment of Pharmaceutical Contaminants to Produce Clean Water for Better Health and Environment. J Cleaner Production, 2023, 387, 135798.
- S.W. Foster, X. Xie, M. Pham, P.A. Peaden, L.M. Patil, L.T. Tolley, P.B. Farnsworth, H.D. Tolley, M.L. Lee, J.P. Grinias. Portable Capillary Liquid Chromatography for Pharmaceutical and Illicit Drug Analysis. J Sep Sci, 2020, 43 (9-10), 1623– 1627.
- D.A.V. Medina, E.V.S. Maciel, A.L. de Toffoli, F.M. Lanças. Miniaturization of Liquid Chromatography Coupled to Mass Spectrometry 2. Achievements on Modern Instrumentation for Miniaturized Liquid Chromatography Coupled to Mass Spectrometry. TrAC Trends in Analytical Chemistry, 2020, 128, 115910.